Copy number pipeline¶
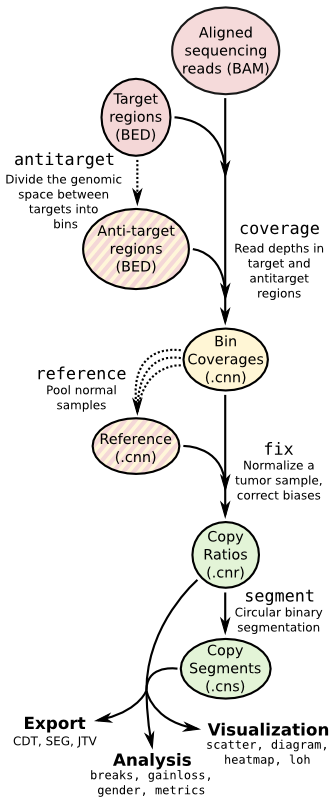
Each operation is invoked as a sub-command of the main script, cnvkit.py.
A listing of all sub-commands can be obtained with cnvkit --help or -h,
and the usage information for each sub-command can be shown with the --help
or -h option after each sub-command name:
cnvkit.py -h
cnvkit.py antitarget -h
A sensible output file name is normally chosen if it isn’t specified, except in
the case of the text reporting commands, which print to standard output by
default, and the matplotlib-based plotting commands (not diagram), which
will display the plots interactively on the screen by default.
batch¶
Run the CNVkit pipeline on one or more BAM files:
cnvkit.py batch Sample.bam -t Tiled.bed -a Background.bed -r Reference.cnn
cnvkit.py batch *.bam --output-dir CNVs/ -t Tiled.bed -a Background.bed -r Reference.cnn
With the -p option, process each of the BAM files in parallel, as separate
subprocesses. The status messages logged to the console will be somewhat
disorderly, but the pipeline will take advantage of multiple CPU cores to
complete sooner.
cnvkit.py batch *.bam -d CNVs/ -t Tiled.bed -a Background.bed -r Reference.cnn -p 8
The pipeline executed by the batch command is equivalent to:
cnvkit.py coverage Sample.bam Tiled.bed -o Sample.targetcoverage.cnn
cnvkit.py coverage Sample.bam Background.bed -o Sample.antitargetcoverage.cnn
cnvkit.py fix Sample.targetcoverage.cnn Sample.antitargetcoverage.cnn Reference_cnn -o Sample.cnr
cnvkit.py segment Sample.cnr -o Sample.cns
See the rest of the commands below to learn about each of these steps and other functionality in CNVkit.
antitarget¶
Derive a background/”antitarget” BED file from a “target” BED file that lists the chromosomal coordinates of the tiled regions used for targeted resequencing.
cnvkit.py antitarget Tiled.bed -g data/access-10000.hg19.bed -o Background.bed
Many fully sequenced genomes, including the human genome, contain large regions
of DNA that are inaccessable to sequencing. (These are mainly the centromeres,
telomeres, and highly repetitive regions.) In the FASTA genome sequence these
regions are filled in with large stretches of N characters. These regions cannot
be mapped by resequencing, so we can avoid them when calculating the antitarget
locations by passing the locations of the accessible sequence regions with the
-g or --access option. These regions are precomputed for the UCSC
reference human genome hg19, and can be computed for other genomes with the
included script genome2access.py.
To use CNVkit on amplicon sequencing data instead of hybrid capture – although this is not recommended – you can exclude all off-target regions from the analysis by passing the target BED file as the “access” file as well:
cnvkit.py antitarget Tiled.bed -g Tiled.bed -o Background.bed
cnvkit.py batch ... -t Tiled.bed -g Tiled.bed ...
This results in empty ”.antitarget.cnn” files which CNVkit will handle safely from version 0.3.4 onward. However, this approach does not collect any copy number information between targeted regions, so it should only be used if you have in fact prepared your samples with a targeted amplicon sequencing protocol.
coverage¶
Calculate coverage in the given regions from BAM read depths.
With the -p option, calculates mean read depth from a pileup; otherwise, counts the number of read start positions in the interval and normalizes to the interval size.
cnvkit.py coverage Sample.bam Tiled.bed -o Sample.targetcoverage.cnn
cnvkit.py coverage Sample.bam Background.bed -o Sample.antitargetcoverage.cnn
About those BAM files:
- The BAM file must be sorted. CNVkit (and most other software) will not notice out if the reads are out of order; it will just ignore the out-of-order reads and the coverages will be zero after a certain point early in the file (e.g. in the middle of chromosome 2). A future release may try to be smarter about this.
- If you’ve prebuilt the index file (.bai), make sure its timestamp is later
than the BAM file’s. CNVkit will automatically index the BAM file if needed
– that is, if the .bai file is missing, or if the timestamp of the .bai
file is older than that of the corresponding .bam file. This is done in case
the BAM file has changed after the index was initially created. (If the index
is wrong, CNVkit will not catch this, and coverages will be mysteriously
truncated to zero after a certain point.) However, if you copy a set of BAM
files and their index files (.bai) together over a network, the smaller .bai
files will typically finish downloading first, and so their timestamp will be
earlier than the corresponding BAM or FASTA file. CNVkit will then consider
the index files to be out of date and will attempt to rebuild them. To prevent
this, use the Unix command
touchto update the timestamp on the index files after all files have been downloaded.
reference¶
Compile a copy-number reference from the given files or directory (containing normal samples). If given a reference genome (-f option), also calculate the GC content of each region.
cnvkit.py reference -o Reference.cnn -f ucsc.hg19.fa *targetcoverage.cnn
If normal samples are not available, it will sometimes work OK to build the
reference from a collection of tumor samples. You can use the scatter command
on the raw .cnn coverage files to help choose samples with relatively
minimal CNVs for use in the reference.
Alternatively, you can create a “flat” reference of neutral copy number (i.e. log2 0.0) for each probe from the target and antitarget interval files. This still computes the GC content of each region if the reference genome is given.
cnvkit.py reference -o FlatReference.cnn -f ucsc.hg19.fa -t Tiled.bed -a Background.bed
Two possible uses for a flat reference:
- Extract copy number information from one or a small number of tumor samples when no suitable reference or set of normal samples is available. The copy number calls will not be as accurate, but large-scale CNVs may still be visible.
- Create a “dummy” reference to use as input to the
batchcommand to process a set of normal samples. Then, create a “real” reference from the resulting*.targetcoverage.cnnand*.antitargetcoverage.cnnfiles, and re-runbatchon a set of tumor samples using this updated reference.
About the FASTA index file:
- As with BAM files, CNVkit will automatically index the FASTA file if the
corresponding .fai file is missing or out of date. If you have copied the
FASTA file and its index together over a network, you may need to use the
touchcommand to update the .fai file’s timestamp so that CNVkit will recognize it as up-to-date.
fix¶
Combine the uncorrected target and antitarget coverage tables (.cnn) and correct for biases in regional coverage and GC content, according to the given reference. Output a table of copy number ratios (.cnr).
cnvkit.py fix Sample.targetcoverage.cnn Sample.antitargetcoverage.cnn Reference.cnn -o Sample.cnr
segment¶
Infer discrete copy number segments from the given coverage table. By default this uses the circular binary segmentation algorithm (CBS), but with the ‘-m haar’ option, the faster but less accurate HaarSeg algorithm can be used instead.
cnvkit.py segment Sample.cnr -o Sample.cns
The output table of copy number segments (.cns) is essentially the same tabular format as the other .cnn and .cnr files.